
Регистрация лекарственных препаратов в России и ЕАЭС
В настоящее время в России вводится ускоренная процедура регистрации лекарственных препаратов направленных на лечение и борьбу с коронавирусной инфекцией COVID-19.
По заявлению регулятора прохождение всех инстанций для регистрации лекарственных средств по займёт 1 неделю.
Производителям будет дано право вносить изменения в инструкцию на лекарственный препарат без проведения исследований.
Планируется заменить лабораторные экспертизы на процедуру, которая действует в режиме гражданской обороны.
Большинство процессов ускоренной регистрации лекарств планируется проводить бесконтактно через электронный кабинет заявителя.
Процедура ускоренной регистрации лекарственных препаратов против коронавирускной инфекции COVID-19 В настоящее время не сформирована.
Схему ускоренной регистрации лекарственных препаратов мы опубликуем в ближайшее время.
Регулируется:
Постановление Правительства РФ от 3 апреля 2020 г. N 441 "Об особенностях обращения лекарственных препаратов для медицинского применения, которые предназначены для применения в условиях угрозы возникновения, возникновения и ликвидации чрезвычайной ситуации и для организации оказания медицинской помощи лицам, пострадавшим в результате чрезвычайных ситуаций, предупреждения чрезвычайных ситуаций, профилактики и лечения заболеваний, представляющих опасность для окружающих, заболеваний и поражений, полученных в результате воздействия неблагоприятных химических, биологических, радиационных факторов"
ГАРАНТ.РУ: http://www.garant.ru/hotlaw/federal/1344231/#ixzz6JapJ883q
На сегодняшний день в России действует только одна схема регистрации лекарственных препаратов:
Регистрация лекарственного препарата в рамках законодательства Евразийского Экономического Союза.
- - Децентрализованная – с одновременным рассмотрением материалов досье всеми государствами-членами ЕАЭС;
- - Централизованная – взаимное признание с последовательным рассмотрением материалов досье каждым из государств.
Для первичного ознакомления с проектом по регистрации лекарства в России нам требуется изучить:
-
Наименование лекарственного препарата (международное непатентованное или химическое и торговое наименование)
-
Состав лекарственного препарата
-
Лекарственная форма, дозировка, способы введения и применения
-
Описание фармакологических и фармакодинамических или иммунобиологических свойств препарата, область применения, форма выпуска
-
В каких странах получено регистрационное удостоверение (Marketing Authorisation)
Схема регистрации лекарственного препарата в референтном государстве
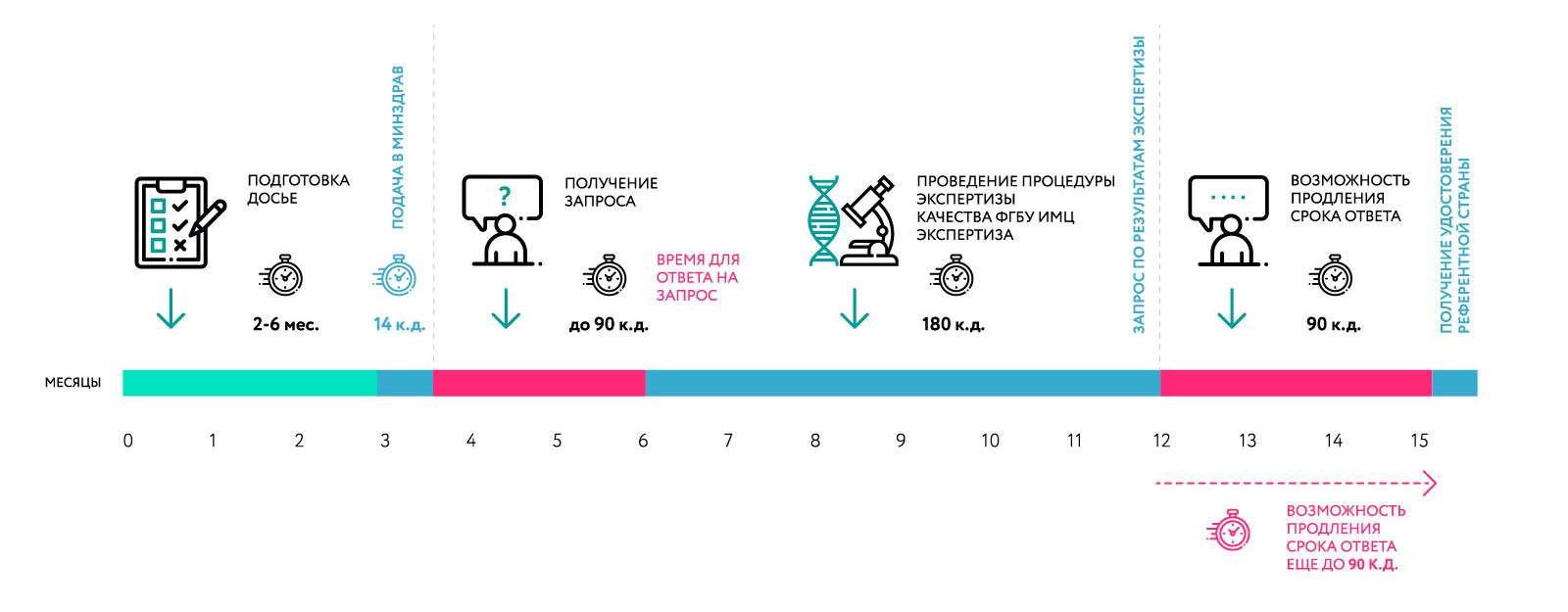
Основные нововведения при регистрации лекарств в рамках процедуры ЕАЭС:
- Необходимость проведения аудита GMP стандарта площадки производителя. подробнее
- Возможность признания клинических исследований, проведённых производителем до 2016 года в странах ICH (Евросоюз, США и Япония) + Швейцария, Канада, Австралия и др. Всего порядка 35 государств.
- Срок регистрации лекарственного препарата 7-9 месяцев.
ВНИМАНИЕ: Все досье на ЛС, которые зарегистрированы по национальным правилам, должны быть приведены в соответствие с законодательством ЕАЭС и поданы для получения нового РУ, действительного на всей территории ЕАЭС до 31 декабря 2025 г.
Каждый проект вывода на рынок лекарственного препарата рассматривается индивидуально.
Последовательность шагов для регистрации лекарственного препарата в Рамках ЕАЭС:
Заключение соглашения о неразглашении конфиденциальной информации и предоставление заказчиком документов для формирования коммерческого предложения.
-
Анализ / формирование Модуль 2 CTD досье. 1-3 месяца
-
Подготовка плана проведения мероприятий, расчет стоимости и сроков. 2 недели.
-
Получение разрешения на клинические исследования. 2 недели
-
Ввоз образцов. 2 недели.
-
Проведение доклинических и клинических исследований (При необходимости) 1-3 года.
-
Одновременно с исследованиями организация выездного аудита GMP стандарта на площадках производителя. 7 месяцев - 1 год.
-
Подача регистрационного досье в Минздрав с результатами исследований и сертификатом GMP. Проведение исследований в рамках регистрации лекарственного препарата. 7-9 месяцев.
-
Принятие решения о включении лекарственного препарата в Государственный реестр лекарственных средств. Выдача свидетельства о государственной регистрации препарата. 1 месяц.
Заявитель самостоятельно выбирает референтное государство и процедуру регистрации лекарства.
До 31.12.2020 г фармацевтические компании смогут выбрать по каким правилам – национальным или единым – будет проходить регистрация ЛП;
В ЕАЭС будут существовать две процедуры регистрации лекарственных средств:
С 01.01.2021 года подать новые ЛС на регистрацию можно будет только по Правилам ЕАЭС (Решение № 78 от 03 ноября 2016 г «О Правилах регистрации и экспертизы ЛС для медицинского применения»):
ЛС может быть зарегистрировано в разных странах Союза, и в зависимости от этого сможет обращаться только в тех странах, где регистрация подтверждена;
Процедура актуализации ранее зарегистрированных лекарств (действует до 31.12.2025)
В России регуляторные функции в фармацевтической сфере исполняются разными государственными ведомствами:
-
Регистрация препаратов – Минздрав
-
Исследования при регистрации лекарственного препарата - ФГБУ ИМЦ «Экспертиза»
-
Лицензирование производства и инспектирование по GMP – Минпромторг
-
Надзор за качеством на рынке, регистрация медицинских изделий (лабораторная служба) – Росздравнадзор
Мы предоставляем услуги по подтверждению регистрации лекарственных средств в странах ЕАЭС.
Наша партнерская сеть: Белоруссия, Казахстан, Узбекистан, Киргизия, Молдавия, Армения, Грузия.